Switzerland’s Medical Device Regulation and the EU Regulations
The medical device industry is thriving globally. Not only in European Union countries, but the influence is also seen in neighboring countries as well. Switzerland, in particular, has become a huge market in medical devices recently.
The Swiss medical device market includes a diverse range of products like imaging systems, surgical instruments, diagnostic tools, implants, and so on. This huge medical device industry is key to Switzerland’s excellent healthcare system as it delivers new solutions to improve patient outcomes and overall healthcare delivery.
However, Switzerland also has its own medical device regulations that control the medical devices sold and distributed in the country. In this article, we will learn about those regulations and how they affect an EU-based medical manufacturer.
Why the Swiss need separate regulatory systems
Switzerland’s cause for having its own medical regulatory system is the same as any other country.
Because medical devices are vital for the diagnosis, treatment, and monitoring of numerous medical diseases, ensuring patient safety and device effectiveness is crucial. And what’s a better way to do it than to have a few sets of rules in place?
Not to mention these restrictions also aid in the maintenance of quality standards and the protection of public health. Meaning that there is a safety net in place if there is ever a public health emergency or mass medical device malfunction (although unlikely).
However, since the EU medical devices regulation is so vigilant when it comes to medical devices, the Swiss authorities accept EU MDR-passed devices (devices that received the CE mark) easily through the mutual recognition agreement.
Nevertheless, although Switzerland is a third country in the EU, its medical device regulation is still active in terms of market surveillance and keeping the existing devices in check. There are a few authorities for keeping Swiss manufacturers in check as well.
Regulatory Landscape in Switzerland
Switzerland regulates medical devices in a similar manner to the EU, which means that the level of regulation is based on the potential dangers or risks connected with a certain item. The categorization method is based on four categories (Class I, IIa, IIb, and III) based on the EU Medical Device Regulation 2017/745 (MDR).
Obviously, Switzerland has its own set of laws. The Swiss Medical Devices Ordinance (MedDO) is the regulatory equivalent of MDR in Switzerland. It also establishes criteria for medical devices not covered by the MDR. These laws encompass topics like design and production controls, clinical trials, labeling, and post-market surveillance.
Overview of Swiss regulatory authorities responsible for medical device regulations
- Swissmedic – Swissmedic stands for the Swiss Agency for Therapeutic Products. Similar to the EU regulatory authorities, Swiss medicine is responsible for regulating all medical devices in Switzerland. When a medical device manufacturer wants to apply to place their device in the Swiss market, they have to go through Swissmedic. They also ensure the medical devices ordinance is followed.
- Federal Office of Public Health (FOPH): The FOPH is associated with public health rather than medical devices. However, some of its work does involve medical devices, especially making sure they comply with health-related requirements, such as efficacy and safety, and providing guidance on reimbursement and pricing.
- International Medical Device Regulators Forum (IMDRF): The IMDRF works in the harmonization of international medical device regulatory laws and regulations. The swissmedic follows the IMDRF regulations, which is part of it, too. So, it’s a good idea to familiarize yourself with these laws, too.
The process of getting approved
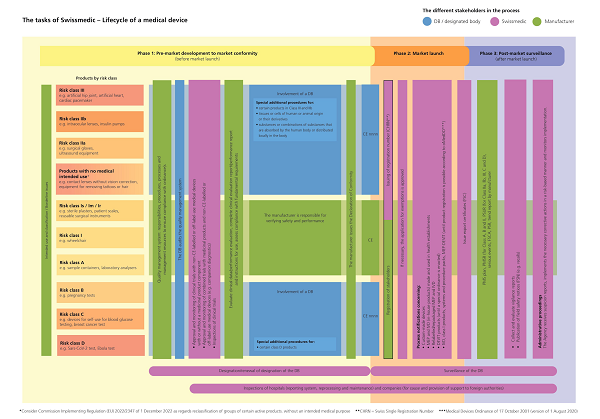
There are three steps or phases of getting a medical device approved in Switzerland.
Pre-market development:
During this phase, the manufacturer is expected to get all their documents ready, the tests and developments done, and all the boxes checked. No worries, the requirements are similar to the CER in EU MDR. This step just means that you have ensured that your medical device meets all necessary requirements and regulations according to its risk category.
The Designated bodies (Swiss equivalent of notified bodies) will give a special focus on the quality management system, so make sure you have it ready.
Market launch:
Once a manufacturer is confident that their device is compliant and the DB clears them, they can submit an application to Swissmedic. Swissmedic will review the application. During this process, there might be some more hoops to jump through; for example, if your device is custom-made or IVD (In Vitro diagnostic medical devices), they might take a deeper look.
Post Market Surveillance
As mentioned earlier, Swissmedic takes PMS seriously. The designated bodies will go through all your PMS data and plans, including PMSR, PSUR, serious event reporting, FSCA, PSR, trend reporting, etc.
Special considerations
High-risk medical devices are always risky to approve. Hence in ,Switzerland high-risk devices such as implantable devices custom made devices require certain conditions and approval processes.
In addition to the regular review process, class III implantable devices must undergo a conformity assessment approved by Swissmedic.
If deemed necessary, clinical trials may be required for any devices the swissmedic requires to demonstrate their safety and effectiveness. These clinical trials will be held under swissmedic inspection.
There will be additional procedures during the designated body approval for devices that use “tissues or cells of human or animal origin or their derivatives or substances or combinations of substances that are absorbed by the human body or distributed locally in the body.”
Custom devices will have to carry a declaration in accordance with Section 1 of Annex XIII to EU-MDR when they are sold.
Timeline and costs
The approval process for medical devices in Switzerland varies depending on the type and complexity of the device. Swissmedic typically takes 6-12 months to approve a medical product.
In terms of expenses, it falls on the manufacturers to pay fees for each stage of the approval procedure, which includes application submission, review, and post-market surveillance.
Getting an exact number is tough because fees can range from a few hundred Swiss Francs to several thousand, depending on the type of equipment and the amount of evaluation necessary.
Our advice to any EU manufacturer looking to launch their product in the Swiss market will be to research and draw up a budget before starting. Hiring a regulatory specialist or even contacting Swissmedic directly could help you get an estimate easily.
FAQ
What are Designated Bodies?
A notified body (EU term) and a designated body (Swiss term) are pretty much the same thing.
Designated bodies, like in the EU, undertake the conformity assessment of all medical devices that aren’t low-risk class I. If everything goes well, they give the device conformity certificate (similar to CE marking). The Swissmedic inspects Swiss designated bodies according to their laws.
