Medical Device Reporting (MDR): How to Report Problems to the FDA
Even though every medical device manufacturer does their utmost to ensure their medical devices are safe and efficient, adverse events still happen. Off-label use, installation/service issues, lack of training, and many other causes can lead to suspected medical device-associated deaths, serious injuries, and malfunctions.
Every year, the Food and Drug Administration (FDA) receives hundreds of thousands of medical device reports from manufacturers, users, importers, healthcare professionals, patients, etc.
The analysis of initial medical device reports (MDR) and subsequent supplemental reports is essential to the FDA’s post-market surveillance responsibilities. It provides critical information regarding the safety and efficiency of medical devices in the United States market.
The medical device reporting (MDR) requirements are described in the FDA Code of Federal Regulations (CFR) Title 21, part 803.
This article will review the medical device reporting (MDR) requirements, important stakeholders, the process required for reporting a medical device problem as a medical device manufacturer or user, and the critical deadlines for medical device reports.
Mandatory medical device reporting requirements
The medical device regulation specifies mandatory medical device reporting (MDR) requirements for manufacturers, importers, and device user facilities and certain device-related adverse events and product problems.
Manufacturers
Manufacturers must report to the FDA if any medical devices have caused or contributed to a death or serious injury. They must also report malfunctions likely to cause or contribute to death or serious injury if they recur.
Importers
Importers are subject to the same reporting responsibilities as the manufacturer. They must report to the FDA and the manufacturer if any of their devices have caused or contributed to a death or serious injury. They must also report any malfunctions that are likely to cause or contribute to death or medical device-related serious injuries if they recur.
Device user facilities
Device user facilities may be hospitals, ambulatory surgical centers, nursing homes, outpatient diagnostic centers, or outpatient treatment centers. These device user facilities must report suspected medical device-related deaths to the FDA and the manufacturer. In the case of serious injury, they must report to the manufacturer or the FDA if the manufacturer is not known.
There are no requirements for device user facilities to report device malfunctions, but they can voluntarily report them to the FDA.

Voluntary malfunction summary reporting program
Medical device manufacturers are permitted to report certain device malfunctions quarterly through the Voluntary Malfunction Summary Reporting (VSMR) program, a part of the medical device regulation established in 2018 in the federal register. A summary report must be submitted for each combination of brand name, device model, and problem code and should identify the total number of reportable malfunctions. The reports are publicly available in the MAUDE database.
Malfunctions not included in the voluntary malfunction summary reporting program
Not all malfunctions for all medical devices are eligible under the voluntary malfunction summary reporting program. There are five types of malfunctions that are mandatory to report:
- Any malfunction associated with a 5-day report, as described in article §803.53(a). 5-day reports under this article include events, or malfunctions, where the action is required to prevent the risk of substantial harm to public health.
- Devices subject to recalls to address malfunctions or malfunctions of the same nature with the same or similar devices.
- Malfunctions that constitute a public health issue. These could be reusable devices and present high risks of infection, malfunctions where the root cause is not well understood, devices under safety-related investigations, etc.
- Malfunctions of devices manufactured by companies that are no longer allowed to participate in the voluntary malfunction summary reporting program. These could be manufacturers with compliance issues or undergoing safety-related investigations.
- New malfunctions that have not previously been reported to the FDA by the manufacturer for that device.
In addition, products included in new product codes with less than two years of existence cannot participate in the program.
Voluntary medical device reporting by the public
The FDA encourages anyone who comes into contact with medical devices and experiences problems, whether health professionals, users, patients, caregivers, or consumers, to submit voluntary reports. Product issues and adverse event reports can be submitted through MedWatch, the FDA’s Safety Information and Adverse Event Reporting Program.
Medical device reporting (MDR): how does it actually work?
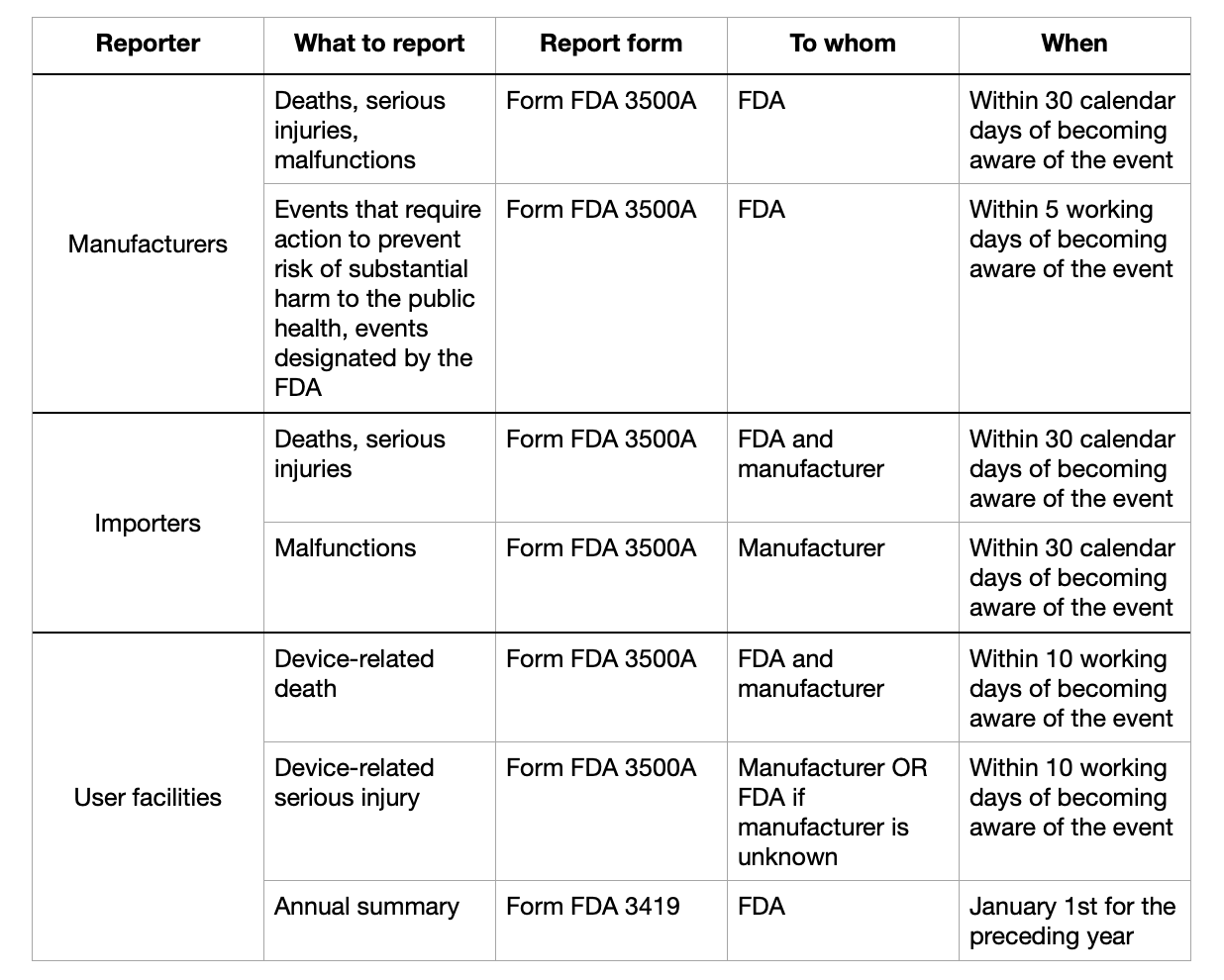
Deadlines
Medical device manufacturers must report any death, serious injury, or malfunction within 30 calendar days of becoming aware of the event.
5-day reports must be submitted within five working days of becoming aware of the event.
Medical device importers must submit reports on death, serious injuries, and malfunctions within 30 calendar days of becoming aware of the event.
Reporting forms
All four reports must be submitted through MedWatch form FDA 3500A. The FDA provides detailed instructions on how to fill out the form. Essential information to include in the form is patient information, product problem or adverse event data, suspect product(s) or medical device(s), and information on the person reporting.
Device user facilities must report suspected medical device-related deaths to both the FDA and manufacturers within ten working days after discovering the event via MedWatch form FDA 3500A, while serious injuries only need to be reported to the manufacturer (or to the FDA is the manufacturer is unknown), also within ten working days. They must also submit yearly reports to the FDA through form FDA3419.
Overview of the reporting process and deadlines
Biological products
Most medical devices are regulated by the Center for Devices and Radiological Health (CDRH); however, biological products are overseen by the Center for Biologics Evaluation and Research (CBER). In vitro diagnostic medical devices for screening or confirmatory clinical laboratory testing associated with blood banking practices and other process testing procedures fall under the CBER.
These IVDs are also included under 21 CFR, part 803, and the medical device reporting (MDR) requirements are mostly the same, as are the forms.
Conclusion
As a medical device manufacturer, it is never pleasant knowing your device is causing people harm. However, it is an inevitable part of producing devices that are part of life-and-death situations. Understanding the reporting process and deadlines when adverse events unavoidably happen is critical for acting swiftly and minimizing harm.
