Clinical Evaluation Report (CER) vs. Periodic Safety Update Report (PSUR)
Every medical device on the European market must be continuously monitored to confirm its safety and efficiency for patients and users. The EU MDR (2017/745) requires clinical evaluation and post-market surveillance programs to ensure the safety of medical devices throughout their lifetime.
The clinical evaluation report and the periodic safety update report are critical documents in demonstrating the continued safety and performance of medical devices, but what is the purpose of each report? And how do they differ?
In this article, we go through the intended purpose and content of each report and explain why both reports are needed under the EU MDR.

Clinical evaluation report (CER)
The Clinical Evaluation Report (CER) is a critical component of the Technical File for conformity assessment and subsequent CE Marking in Europe.
It is an evaluation and conclusion of the clinical data collected for your device with the specific purpose of demonstrating that your medical device is safe and efficient while delivering on clinical performance. More specifically, a Clinical Evaluation Report documents the assessment and analysis of clinical data collected for a medical device to verify the clinical safety and efficacy of the device.
It is comprised of a clinical investigation of the device itself and/or existing clinical studies for equivalent devices, as described in article 61 of the MDR:
“3. A clinical evaluation shall follow a defined and methodologically sound procedure based on the following:
a) a critical evaluation of the relevant scientific literature currently available relating to the safety, performance, design characteristics, and intended purpose of the device, …
b) a critical evaluation of the results of all available clinical investigations, …
c) a consideration of currently available alternative treatments options for that purpose, if any.”

Considering the many different types of medical devices out there, it is difficult to write a definitive list of what should be included in the clinical evaluation report. As you can imagine, higher-risk devices can significantly impact a patient’s health in case of malfunction and will require a more detailed clinical evaluation report than lower-risk devices.
Even though the detail required in the report varies greatly, the general structure of the clinical evaluation report remains the same.
You should include the following elements:
- Device information, such as intended use, medical device design, manufacturer name, regulatory history, and any other relevant data.
- Technical device description, such as intended use and target population, classification, therapeutic or diagnostic claims, clinical background, etc.
- Any identified clinically equivalent device and a justification of its equivalence
- Existing clinical investigation and data
- Summary of clinical data and review, including documented systematic literature searches and reviews
- Data appraisal methodology and parameters
- Conclusions about safety, performance, and conformity, as well as benefit-risk determination
Periodic safety update report (PSUR)
Periodic safety update reports are well-known in the pharmaceutical world, but a new requirement in the medical device industry.
When the EU MDR was being developed, the medical device industry expected a stricter focus on post-market surveillance data systems due to the emergence of several unfortunate cases with medical devices that had not been monitored sufficiently. And as expected, the MDR puts a much greater emphasis on safety throughout the lifetime of the medical device – post-market surveillance is now a focal point in the regulation and a critical part of the obligations of the medical device manufacturer.
The periodic safety update report is described as a mandatory requirement per article 86 of the MDR:
“Throughout the lifetime of the device concerned, the PSUR shall set out:
a) the conclusions of the benefit-risk determination;
b) the main findings of the PMCF; and
c) the volume of sales of the device and an estimate evaluation of the size and other characteristics of the population using the device and, where practicable, the usage frequency of the device.”
The report should be continuously updated throughout the lifetime of the device once the device is put on the market. The periodic safety update report is a part of the technical documentation submitted to the notified body during the conformity assessment.

The periodic safety update report is not merely a repetition of the post-market surveillance report, but a separate document underlining the safety of the medical device in question.
The report should generally include the following elements:
- Conclusions on benefit-risk analysis
- Findings from the post-market clinical follow-up
- Sales volume and usage frequency of the device
- Vigilance data
- Descriptions and rationales for any preventive and/or corrective actions made
- Results and conclusions on the post-market surveillance data
So, what are the differences between the CER and PSUR?
While there is some overlap between the two reports, they do not serve the same purpose.
The clinical evaluation report demonstrates safety and efficiency for a medical device’s clinical use, before and after product launch, interpreting all the clinical data found on the device.
The periodic safety update report only concerns itself with a demonstration of safety and performance based on post-market data.
The periodic safety update report also helps manufacturers demonstrate to the notified body that their post-market surveillance program uses the data available on the market to ensure the continued safety, efficacy, and performance of their device – It proves the efficacy of a proactive post-market surveillance system.
Below you will find a summary of the elements included in each report.
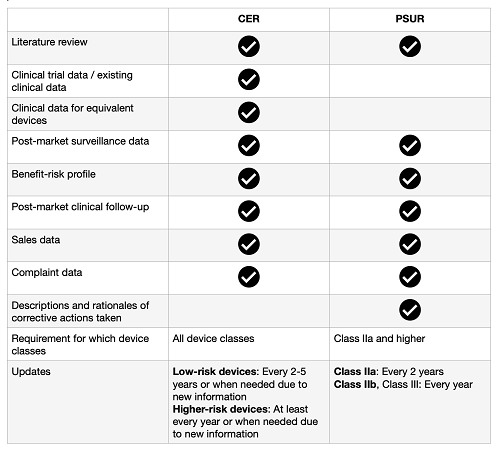
The implementation of the EU MDR in May 2021 and its emphasized focus on efficient clinical evaluation and post-market surveillance systems has put a lot of pressure on medical device manufacturers. Regulatory staff are struggling under the new requirements and the time pressures of getting clinical and post-market surveillance reports up to date – and keeping them updated.
The notified bodies will be vigilant in their reviews of these reports and ensuring compliance to clinical and post-market requirements will become a greater part of the regulatory work related to medical devices post-launch.
If your team is struggling, CiteMed can help with the clinical evaluation report and post-market surveillance requirements for your medical device. Let’s talk!
