CER for Software as a Medical Device in the EU
The medical device industry has been changing bit by bit every day, and one of the significant changes is the addition of technology to medical devices. While medical testing devices, scans, and robotic surgical instruments were already in use, they were mostly manual and user dependent. The use of the software is not new but certainly not very old.
Software used in conjunction with medical devices has become increasingly important over recent years, with many new applications developed to improve patient care in both hospitals and community settings.
Like any other medical device, the safe and effective use of the software is vital for patients and healthcare professionals alike.
Since this software is technically being used similarly to a medical device, the MDR sets out the requirements for ensuring their safety, efficacy, and quality for them, along with other medical devices.
What exactly is Software as a medical device?
Software as a medical device is software that performs or supports the functions of a medical device. This software may be embedded in a device, such as a pacemaker, or it may be used in conjunction with the device, such as an app for an iPhone that analyses and records data from the heart.
Even if it is a part of a medical device, the software has to have its own medical function to qualify as a medical device.
The MDR sets some criteria:
- The software has to be considered as software according to “MDCG 2019-11”.
- The software can work independently or in conjunction with a medical device but should have its own effect to be considered a medical device.
- The software can be operated from anywhere, whether attached to the device, phone, or cloud.
- The software can be intended to be used by healthcare personnel or layperson.
- Even if the software is an accessory or doesn’t have any medical use independently, it falls under the jurisdiction of MDR.
- The details of the software and accessory can be found in the ‘MDR Annex XVI device and ‘Accessory’ in the Art. 2(2) of the MDR or IVDR.
This software can include any type of computer program code (e.g., operating systems), firmware, etc. It also includes any information recorded on material media intended for use in digital devices intended for medical purposes. However, software that is not necessarily clinical or related to the patient directly, for example, payment-related software, is not considered SaMD.
The In vitro diagnostic device software devices are covered by the REGULATION (EU) 2017/746 (IVDR). SaMD, which works exclusively from data sourced by IVD devices, is considered within these regulations. Others fall under (EU) regulation 2017/745 or MDR.
The software that isn’t considered SaMD are:
- Software that only works with data for storage, archival, communication, or simply search.
- It has no function to benefit individual patients.
- And finally, if it isn’t considered medical device software (MDSW) according to the definition of MDCG 2019-11.
MDSW Classification
The MDR classifies the medical device software into four categories: Class I (low risk), Class IIa (medium risk), Class IIb (medium/high risk), and Class III (high risk). Based on the categories, the requirements for clinical evaluation and performance evaluation reports also vary.
Softwares that factor in the physician’s decision-making regarding diagnosis or therapeutic purposes are considered class IIa. Software that works as a physiological processes monitor is classified as class IIa. The vital parameter monitors are considered class IIb.
Except for a few exclusion criteria, all other medical software is considered class I.
UDI assignment

Each medical device software has to be assigned a UDI. Usually, it’s given on the system level of the software, as they are part of medical devices. The software that is individually commercially available will need a UDI assignment on its own.
The UDI is part of the manufacturing control mechanism, and the UDI-PI will display it. Major changes in the software like:
- Changes in the original performance.
- Changes in the safety or the intended purpose
- Changes in data interpretation.
Other minor changes will need an update in the UDI-PI.
CER for software as a medical Devices
The clinical evaluation process for medical device software is pretty similar to other medical devices. Most of the medical device software is not high-risk class, so the requirements are, in fact, a bit lenient than high-risk devices. However, the notified body is involved.
On the MDCG 2020-1 Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software document, the Medical Device Regulation provided the following chart as the standard medical device clinical evaluation to-do for software as a medical device.
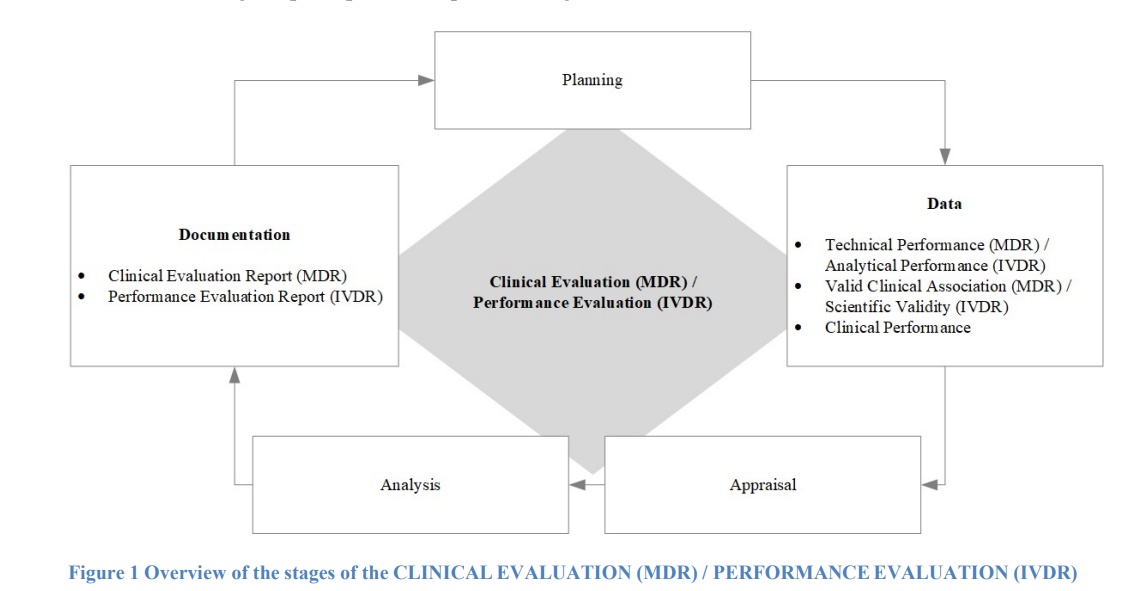
The key finding from this document for medical device manufacturers are:
- All requirements of CER and PER can be found in Article 61 of the Medical Device Regulation (and Annex XIV) and Article 56 of IVDR (and Annex XIII).
- The clinical evidence must be sufficient and appropriate based on the device’s properties, risks, and intended use.
- The clinical situation, condition, indication, or parameter of the device’s intended use has to be proven with sufficient evidence for scientific validity or valid clinical association. In short, the device has to have a clear, exact clinical use.
- The device’s technical performance or analytical performance has to be proven by creating correct output based on the input.
- Its clinical performance has to be proven by producing correct, clinically evidence-relevant output that has a positive impact.
- If the device’s clinical benefits are not obvious, it can pass using its usability.
- The device must also have a valid risk-benefit ratio compared to the state of the art.
- The above-mentioned evidence will act as the clinical evidence, which needs to be of sufficient amount and quality.
- Clinical data of several parameters and scopes must be submitted for the device to be considered for adequate clinical performance. The difference for medical device software is that since they are modular, the testing can be done in modular form as well.
- For the software that falls under IVDR, clinical performance studies are a must.
- Apart from all these, unless the requirements of Article 61(4), (5), or (6) of the MDR are fulfilled, a clinical investigation is needed.
- The clinical investigation or clinical performance study is usually ordered to ensure safe and effective software. It can be a prospective or retrospective study based on the software’s use.
- After all the documents have been collected, the risk-benefit analysis is done the clinical evaluation is ready for submission.
Do I need a checklist?

The CER of software as a medical device and regular medical devices are similar in their complexity, so we would suggest starting as prepared as you can. A checklist is always a great tool to have for streamlining the process. It will also ensure you do not forget all the small details required by software as a medical device.
Further Reading
- Microsoft Word – Clinical Evaluation l Performance Evaluation of Medical Device Software – website.docx (europa.eu)
- EUR-Lex – 32017R0745 – EN – EUR-Lex (europa.eu)
