Post-Market Surveillance and Vigilance: MDR vs IVDR
The Medical Device Regulation 2017/745 (MDR) and the In Vitro Diagnostic Regulation 2017/746 (IVDR) are two sides of the same regulatory coin in almost all aspects: both regulations seek to set up a regulatory system that will increase safety and efficiency in the European medical device market; both include collecting clinical evidence and demonstrating an acceptable risk-benefit profile of medical devices, and; both set up strict post-market surveillance and vigilance requirements.
The IVDR has been harmonized with the MDR – particularly in regard to supply chain, clinical evaluations, and post-market requirements – compared to the In Vitro Diagnostic Device Directive (IVDD) that came before, making it easier for manufacturers to understand and comply with both regulations.
Although the general content of the two regulations is much the same, there are a few differences.
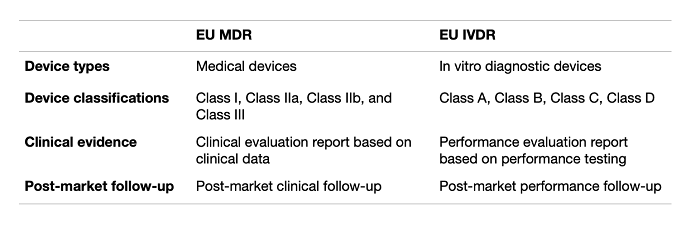
Post-Market Surveillance and vigilance in the Medical Device Regulation
Post Market Surveillance (PMS) is defined in the European medical device regulation as “all activities carried out by the manufacturer… to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market”.

It is the single most important tool in medical device manufacturers’ toolbox to analyze and document any and all adverse events happening in situations where devices are included, and it is a mandatory process under practically every medical device regulation in the world.
Post Market Surveillance Reports (PMSR) and Periodic Safety Update Reports (PSUR)
One of the new initiatives in the MDR compared to the MDD is the requirement to prepare Post Market Surveillance Reports (PMSR) and Periodic Safety Update Reports (PSUR) for medical devices – This requirement has previously only been present in the pharmaceutical world.
Of these two reports, the PMSR applies only to low-risk Class I devices and should only be provided to the authorities on request. The PSUR applies to Class IIa, Class IIb, and Class III devices and should generally be updated at least annually.
Post Market Clinical Follow-up (PMCF)
Besides the PMSR and PSUR reports, a Post Market Clinical Follow-up (PMCF) report should also be elaborated as part of the concept of continuous clinical evaluation and lifecycle management of your medical device. The PMCF requirements are stricter and more intense than under the MDD and the guidance for PMCF has been reinforced and expanded.

Vigilance reporting
Last, but not least, vigilance reporting must be a strong component of any Post Market Surveillance program and medical device vigilance system. When actions are taken to reduce the risk of harm and/or serious deterioration of health by the manufacturer as part of their adverse event reporting program, it is the responsibility of the manufacturer (or the Authorized Representative) to report these actions to the competent authorities in the afflicted markets. Failure to report a Field Safety Corrective Action and present vigilance reports can result in serious legal consequences.
Post-Market Surveillance and vigilance in the In Vitro Diagnostic Medical Device Regulation
Post Market Surveillance (PMS) is defined in the European in vitro diagnostic device regulation almost identically as in the MDR. Post-market surveillance is defined as “all activities carried out by the manufacturer… to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions.”

Post Market Surveillance Reports (PMSR) and Periodic Safety Update Reports (PSUR)
As in the MDR, the IVDR demands Post Market Surveillance Reports (PMSR) for all Class A and B IVDs, and Periodic Safety Update Reports (PSUR) for Class C and D IVDs.
Post Market Clinical Follow-up (PMCF)
The clinical evaluation report – so well-known for medical devices – should be substituted for a performance evaluation report for IVDs, as clinical evidence for in vitro diagnostic medical devices is demonstrated through performance evaluations, not clinical evaluations. The post-market clinical follow-up for IVDs is also substituted for a post-market performance follow-up report (PMPF).
Since the risk of in vitro diagnostic medical devices depends so much on the device type, the post-market surveillance requirements are not as defined as they are for medical devices – a Class A IVD will require different post-market surveillance activities than a Class D IVD.

Vigilance reporting
Vigilance activities work much the same for IVDs as they do for medical devices – the manufacturer must notify the national competent authority of any actions taken to reduce the risk of harm and/or serious deterioration of health, such as field safety corrective actions, within the established timelines.
So, what are the actual differences in regard to post-market surveillance and vigilance in the MDR vs IVDR?
The short answer is that there are not any – both regulations require robust initiative-taking post-market surveillance and vigilance systems and mention the same deadlines and timelines for post-market surveillance reports, periodic safety update reports, post-market clinical/performance follow-up, and vigilance reporting.
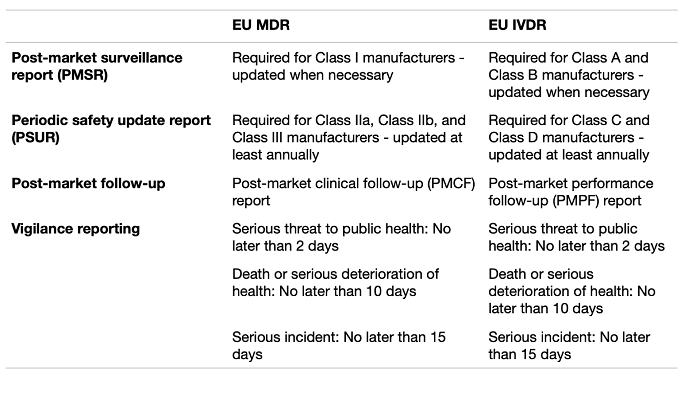
The differences can be found in the minor details of the post-market surveillance and post-market follow-up requirements, such as the specific content of the reports, as this depends on your device type and risk classification.
At this point in time, the major difference between the MDR and the IVDR is the roll-out of each regulation. The MDR came into force in 2021, while the IVDR is set to begin implementation on May 26th, 2022.
Also, the certification of notified bodies to manage demands for conformity assessment for IVDs is struggling to keep up with medical device notified bodies. Currently, we have 28 notified bodies certified to MDR and only 7 notified bodies certified to IVDs.

As a manufacturer of medical devices and in vitro diagnostic devices, you will find the MDR and IVDR harmonized and similar in regard to post-market surveillance and vigilance requirements. The differences lie in the subtleties, so if you manufacture both types of devices you will need to study the two regulations carefully to point out the differences and comply with both regulations.
